This is a selection of patient histories that appeared in my book The Sicklecell Disease Patient [1996]
1. Osteomyelitis in Sickle Cell Disease Patients
On 22nd September 2011 the Senior Resident of a well known Teaching Hospital writes to me about a chronically ill sickle cell disease patient. He asks whether the limb deformities he describes fit a diagnosis that I am familiar with, and whether he needs to invoke additional pathology. As I had described similar conditions 20 years ago in my book “The Sickle Cell Disease Patient” I think the time has come to present these from my archives for wider readership. From my 133 “Illustrative Case Histories” on the Sickle Cell Disease Patient I begin with what the Senior Resident has asked my opinion about. I reproduce verbatim the entry on pages 477 and 478 of my book.
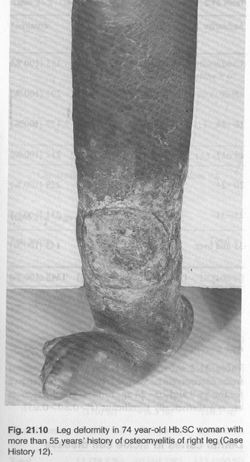
Case History 12: To illustrate the mutilating effect of chronic osteomyelitis in long surviving patients. More than 55-year history of osteomyelitis in Sickle Cell Haemoglobin C Disease.
K. H. (Serial Number 1116). Female aged 74 years. This literate woman, born in 1896 and educated by the Basel missionaries, had been rheumatic since childhood. In her own words: “As soon as it rains, I get the trouble”. Discharging sinuses on right foot and leg, left arm, and spine began in 1915. The leg sinuses continued unabated until the whole foot became reduced to a diminutive stump [Fig. 21.10 see picture below], when seen in March 1969. Pale and jaundiced, she had a liver 6 cm below the costal margin, and tender. Spleen was not palpable. Pulse 80 /min; BP 110/70. There was inguinal lymphadenopathy. There were scars of healed osteomyelitis on left back and left arm. Right leg ulcerated with brawny oedema. Right foot disfigured and reduced in size [Fig. 21.10]. Bilateral incipient cataract with left retinitis proliferans and colloid bodies. Urine, albuminuria 2+ but amyloidosis not confirmed. Hb 8.0 g %, WBC 7500/mm3 with N 70%, L 25%. Serum proteins 6.8 g% (alb 3.1, glob 3.7%). Bilirubin 1.1 mg % (dir. 0.3 mg, ind 0.8 mg %). Wound swab, E. coli and Staph pyogenes.
Comment:. Sickle cell disease must be suspected in any African patient with gross joint or bone deformity of long standing especially if there is a history of discharging sinuses which point to chronic osteomyelitis. This and other “illustrative cases” are to be found in Konotey-Ahulu FID. “The Sickle Cell Disease Patient: Natural History from a clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic” Published by The Macmillan Press Ltd., London and Basingstoke 1991&1992 Reprinted by T-A’D Co, Watford UK, 1996 ISBN 0-9515442-2-5 [The 643-page book can be purchased from this website]
2. Pregnancy in Sickle Cell Disease Patients
On Thursday 22nd September 2011 BBC’s Radio 4 in the UK had on its Women’s Hour a programme on Sickle Cell Disease and Pregnancy. Listening to it, a Ghanaian mother of two adult sickle cell anaemia ‘Patient Achievers’ was upset by what she thought was a piece of “misinformation” in the programme. The doctor leading the interview of an African mother who had just gone through pregnancy in London (one Dr Joe Howard of Guy’s/St Thomas’ Teaching Hospitals Group) seemed to imply that pregnancy by sickle cell disease patients was so hazardous that, in developing countries especially, a complication such as eclampsia with swollen feet and high blood pressure is to be expected. The Case Histories of more than 40 years ago tell a different story.
My own archives show that pregnancy outcome, like the outcome of the management of any patient in general, depends on the carer. Ghanaian midwives know this hereditary disease (chwechweechwe/ahotutuo/nwiiwii) and are capable of successfully supervising many pregnancies in the same patient. The world records were held by Ghanaian women one of whose exploits were published in the British Medical Journal [see Case 112 below]. My book has a whole chapter on pregnancy in sickle cell disease.
Case History 111. To illustrate more than ten successful pregnancies in Sickle Cell Haemoglobin C woman without a single blood transfusion. Mild EEG abnormality.
A.W. (Serial number 413). This 42-year old Hb SC woman whose children were alive and well out of eleven births had no difficulty during pregnancy itself but as soon as she delivered, and for a fortnight afterwards she often had severe and bone and joint pains. …She agreed to sterilization after her eleventh pregnancy. [See account of her physical signs including sickle retinopathy on pages 512 & 513 of my book].
Case History 112. To illustrate the effect of marriage between sickle cell disease patients. An SC man married to Sbeta-Thalassaemia woman.
The D. Family. The first of this remarkable family to be seen was S. D., now (in 1970) a 20 year old man (serial number 538). Complained of cold season rheumatism. That the whole family was thoroughly investigated and reported (Konotey-Ahulu 1969) was due to his father’s statement: “I myself suffer from nwiiwii and my wife also has nwiiwii, (The name of sickle cell disease in Fante is nwiiwii). The man was Hb SC, the woman Hb Sbeta-Thal. They had 13 children from twelve pregnancies; four of the children are dead, but all the rest have haemoglobinopathy – four SS, 2 SC, two Sbeta-Thal, and one Cbeta-Thal (Figs. 3.2, 27.2, 34.2) and Cover photograph). Comment. Clinicians in Ghana have come to learn that when anyone claims to have sickle cell disease, using the tribal vernacular names, he is always correct.
Page 762 27 September 1969 Medical Memoranda
Thirteen Children from Twelve
Pregnancies in Sickle-cell Thalassaemia**
British Medical Journal, 1969, 3, 762
CASE REPORT
A Fante housewife aged 40 years, known to have sickle-cell
thalassaemia and to have a husband with haemoglobin SC disease,
presented in May 1968 with amenorrhoea of three to four months'
duration. The details of 11 previous pregnancies and the haematological
and haemoglobin electrophoretic findings in her family are
fully reported elsewhere (Konotey-Ahulu and Ringelhann, 1969).
Examination showed her to be well nourished but clinically
anaemic. There was pronounced frontal bossing of her skull and
a large diffuse non-toxic goitre was noted. The pulse and blood
pressure were normal, but a precordial systolic bruit was present.
On palpation of the abdomen the liver and spleen were not felt;
there was enlargement of the uterus compatible with a pregnancy
of 14 to 16 weeks. The haemoglobin level was 9-2 g./100 ml.,
the reticulocyte count 6 %, and the sickling test positive. The
electrophoretic pattern was that of sickle-cell thalassaemia.
The patient was followed up fortnightly until November, when
she was admitted to hospital with a suspected loss of liquor, but
this was not confirmed. She had been well throughout the pregnancy
except for occasional " rheumatic" pains. Labour began
spontaneously on 25 November and she was delivered of a healthy
8-lb. (3,620-g.) male infant. The placenta (see Fig.) was described
The placenta, showing pale areas of infarction.
as "unhealthy-looking," with ragged incomplete membranes and
areas of infarction. There was excessive postpartum blood loss,
for which she received three units of blood.
At the age of 4 months the baby was found to have sickle-cell
thalassaemia. Contraceptive advice was earlier refused by the
mother; later ishe agreed to use an intrauterine device.
COMMENT
Although sickle-cell anaemia and its variants do not always
present a great obstetric problem as in this patient, there are
risks to both mother and child. Curzen (1964) described a case
of " leather pouch " uterus due to extensive myometrial necrosis
in a second pregnancy associated with sickle-cell anaemia. …..
We believe that the patient whose case is reported here might have
died outside hospital because blood would not have been readily
available after delivery of her thirteenth child.
More and more haemoglobinopathy patients are passing
through pregnancy (see Table); we therefore stress the need
for contraceptive measures for them, not only because of the
hazards of pregnancy (Bannerman and White, 1957; Smith
and Conley, 1957; Hook and Cooper, 1958; Smith and
Krevans, 1959) but also to prevent dissemination of the genes.
Number of Patients with Various Sickle-cell Genotypes Followed up
Through Pregnancy at Korle Bu Teaching Hospital (1964-8)
Year SS SC S-Thal. SF SKorle Bu Total
1964 1 8 - - - 9
1965 1 11 - - 1* 13
1966 2 6 - - - 8
1967 2 7 1 1 - 11
1968 3 16 2 1 1 23
Total 9 48 3 2 2 64
* In retrospect.
This is the first recorded case of 12 pregnancies with 13 children
(9 alive) in sickle-cell beta-thalassaemia. The other related
facts of a husband with haemoglobin SC disease and hence the
completely haemoglobinopathic offspring also constitute a world record.
[SF means Sickle cell Hereditary Persistence of Fetal Haemoglobin]
We are grateful to Dr. F. S. Boi-Doku, consultant haematologist,
for the blood profiles, and to Professor Bela Ringelhann for the
Hb electrophoretic findings.
K. K. BENTSI-ENCHILL, B.A., M.B., M.R.C.O.G.,
Professor of Obstetrics and Gynaecology, Ghana Medical
School, Accra.
F. I. D. KONOTEY-AHULU, M.B., M.R.C.P.(LOND., GLASG.)
D.T.M.&H.,
Lecturer, Department of Medicine and Therapeutics,
Ghana Medical School, Accra ; Physician, Korle Bu
Teaching Hospital, Accra.t
In receipt of a grant from International Atomic Energy Agency.
REFERENCES
Bannerman, R. H. O., and White, J. C. (1957). Journal of Obstetrics
and Gynaecology of the British Commonwealth, 64, 682.
Curzen, P. (1964). British Medical 7ournal, 1, 1360.
Hook, E. W., and Cooper, G. R. (1958). Southern Medical 7ournal, 51, 610.
Konotey-Ahulu, F. I. D., and Korsah, K. K. (1964). Unpublished observations.
Konotey-Ahulu, F. I. D., and Ringelhann, B. (1969). British Medical Journal, 1, 607-612.
Smith E. W., and Conley, C. L. (1957). Conference on Hemoglobins, National Research Council, Publication No. 557, p. 276. Washington.
Smith, E. W., and Krevans, J. R. (1959). Bulletin of the 7ohns Hopkins Hospital, 104, 17.
Woodruff, A. W. (1961). In Recent Advances in Tropical Medicine, edited by N. Fairley, p. 415. London, Churchill.
** Presented at a Conference of the Association of Physicians of West Africa in April 1969 at Accra, Ghana
===================================================================
[This and other “illustrative cases” are to be found in Konotey-Ahulu FID. “The Sickle Cell Disease Patient: Natural History from a clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic” Published by The Macmillan Press Ltd., London and Basingstoke 1991&1992 Reprinted by T-A’D Co, Watford UK, 1996 ISBN 0-9515442-2-5 [The 643-page book can be purchased from this website]
DISCLAIMER: Advice given here is what I have given to my patients. It is not meant to supplant that given by other health workers to their patients. Although I imply no criticism in these columns, that does not mean I have not disagreed with others in my peer-reviewed published articles listed in the Publications Section of this Web Site. F I D KONOTEY-AHULU



